

Abstract
Experimental Design: Isobolographic method was used to calculate combination index values from cell viability data. Apoptosis was evaluated in A549 cells by terminal deoxynucleotidyl transferase-mediated nick end labeling assay and measurement of cleaved poly(ADP-ribose) polymerase level. Expression of proteins was studied by Western blotting. A549 cells were implanted to induce orthotopic lung tumors in nude mice and the efficacy of docetaxel, DIM-C-pPhC6H5, or combination was determined. Apoptosis and cleaved caspase-3 expression in the harvested tissues were studied by terminal deoxynucleotidyl transferase-mediated nick end labeling and immunohistochemistry, respectively.
Results: The combination index values (0.36-0.9) suggested synergistic to additive effects of docetaxel + DIM-C-pPhC6H5 and resulted in the highest increase in percentage of apoptotic cells and expression of cleaved poly(ADP-ribose) polymerase, Bax, and N-cadherin compared with treatment with either agent. The combination also enhanced procaspase-3 and -9 cleavage. In vivo, docetaxel + DIM-C-pPhC6H5 reduced lung weights by 57% compared with 39% by docetaxel or 22% by DIM-C-pPhC6H5 alone, induced apoptosis in 43% of the tumor cells compared with 29% and 22% in tumors treated with docetaxel and DIM-C-pPhC6H5, respectively, and increased procaspase-3 cleavage compared with either agent alone.
Conclusions: These findings suggest potential benefit for use of docetaxel and DIM-C-pPhC6H5 combination in lung cancer treatment.
Docetaxel, a taxane anticancer agent, is commonly used to treat several types of solid tumors such as breast, prostate, and lung cancers. Because the high toxic doses currently administered cause unfavorable side effects and the prognosis for (NSCLC) patients remains poor, there is a need to develop alternative treatments with improved clinical outcomes. Therefore, we adopted the combination therapy strategy with the aim of achieving a chemotherapeutic efficacy superior to that of docetaxel alone. We chose DIM-C-pPhC6H5, a PPARγ agonist that specifically targets cancer cells and inhibits tumor growth with minimal or no toxicity in several cancer animal models. Our in vitro and in vivo studies with A549 lung cancer models show for the first time that docetaxel + DIM-C-pPhC6H5 therapy provides better efficacy than therapy with docetaxel alone. Therefore, the findings suggest that the combined use of docetaxel and DIM-C-pPhC6H5 may be of clinical benefit to the NSCLC patient. The optimal therapy will combine intravenous docetaxel at a dose lower than the currently administered dose with a high dose of oral DIM-C-pPhC6H5 for better tumor growth control and side effect profile.
Lung cancer remains the most common cancer worldwide with only 13% of patients surviving >5 years following diagnosis. In the United States alone, in 2006, there were an estimated 162,460 deaths resulting from lung cancer accounting for 29% of all cancer deaths (1). Response and remission rates in non-small cell lung cancer (NSCLC) patients remain relatively low despite advances in lung cancer treatment (2). To address this problem, attention has been focused on finding novel combinations of anticancer agents with nonoverlapping mechanisms of action to achieve enhanced anticancer efficacy with decreased adverse side effects. The use of peroxisome proliferator-activated receptor γ (PPARγ) agonists in combination with taxane exhibited synergistic antitumor effects in breast (3) and additive antitumor effects in thyroid carcinoma (4) cancer models.
PPARγ is a member of the nuclear hormone receptors superfamily of ligand-activated transcription factors, which include receptors for steroids, thyroid hormone, vitamin D, and retinoic acid (5). PPARγ functions as an important regulator of lipid and glucose metabolism, adipocyte differentiation, and energy homeostasis (6–10). This receptor is commonly overexpressed in human primary tumors of the lung, breast, colon and pancreas (8, 11), suggesting that high PPARγ expression may serve as a potential tumor marker. In the lungs, PPARγ is expressed in type II pneumocytes that serve as progenitor cells for the lung alveolar epithelium after injury or during carcinogenesis (12).
PPARγ agonists induce growth arrest and changes associated with differentiation and apoptosis in breast (13, 14), colon (15, 16), prostate (17), and NSCLC (11, 18, 19) cancers. Satoh et al. showed activation of PPARγ by the thiazolidinedione class of antidiabetic drugs, which elicited growth inhibition and induced apoptosis of NSCLC cells (20). The thiazolidinedione ciglitizone exhibited antiproliferative activity in A549, H157, H1334, H522, H322, H522, H1944, and H1299 lung cancer cells (11).
A series of 1,1-bis (3′-indolyl)-1-(p-substituted phenyl) methanes [C-substituted diindolylmethanes (C-DIM)] are a new class of PPARγ agonists (13). Compounds containing p-CF3 (DIM-C-pPhCF3), p-t-butyl (DIM-C-pPhtBu), and p-phenyl (DIM-C-pPhC6H5) are potent activators of PPARγ and inhibitors of cancer cell growth. These C-DIMs induce responses also observed for prostaglandin J2 and other PPARγ agonists and these include apoptosis, inhibition of cancer cell growth, and inhibition of G0-G1-S-phase progression. The three PPARγ-active C-DIMs inhibit growth of colon (21–23), bladder, prostate (24), and breast (13, 25) cancer cells. PPARγ-dependent responses include activation of p21 in Panc-28 pancreatic cancer cells (26) and in induction of caveolins in HCT-15 and HT-29 colon cancer cells at low drug concentrations (22). Moreover, like other PPARγ agonists, C-DIMs induced PPARγ-independent growth inhibition and proapoptotic responses, including induction of p27, activating transcription factor 3, and nonsteroidal anti-inflammatory drug-activated gene-1, and down-regulation of cyclin D1 and caveolin-1 in LNCaP prostate cancer cells (24). Also, activation of markers of endoplasmic reticulum stress and the extrinsic apoptosis pathway was observed in pancreatic cancer cells treated with DIM and C-DIMs and these effects were also receptor-independent (27).
Docetaxel has been approved for the treatment of NSCLC patients, and in lung cancer cells, docetaxel inhibits cell growth and induces apoptosis by stabilization of microtubules. Several researchers have studied the combination of docetaxel and other agents for the treatment of lung cancer (28–32) and reported enhanced anticancer effects. Recent studies in our laboratory have shown that PPARγ agonist prostaglandin J2 in combination with docetaxel exhibits a more pronounced inhibition of tumor growth compared with treatments with either compound alone in nude mice bearing A549 and H460 tumors (33).
In the current study, we have evaluated a model C-DIM compound either alone or in combination with docetaxel in the treatment of lung cancer and have hypothesized that combination treatment of DIM-C-pPhC6H5 + docetaxel may produce additive/synergistic cytotoxic effects in human lung cancer cells in vitro and in vivo possibly by enhancing apoptosis. This is the first report on the activity of the C-DIM compounds alone or in combination with docetaxel against lung cancer (in vitro and in vivo). Therefore, the objectives of this study were to (a) examine the in vitro cytotoxicity of C-DIMs alone and in combination with docetaxel against A549 and H460 cells, (b) study the effects of combined treatment of DIM-C-pPhC6H5 and docetaxel and the compounds alone on apoptosis in A549 cells, and (c) evaluate the antitumorigenic effect of DIM-C-pPhC6H5 alone and in combination with docetaxel in vivo in mice bearing A549 orthotopic lung tumors.
Materials and Methods
Materials. The C-DIMs were synthesized as described previously (13). Docetaxel was a gift from Aventis. The human NSCLC cell lines H460 and A549 were obtained from the American Type Culture Collection. H460 cells were grown in RPMI (Sigma) supplemented with 10% fetal bovine serum. A549 cells were grown in F12K medium (Sigma) supplemented with 10% fetal bovine serum. All tissue culture media contained antibiotic/antimycotic solution of penicillin (5,000 units/mL), streptomycin (0.1 mg/mL), and neomycin (0.2 mg/mL). The cells were maintained at 37°C in the presence of 5% CO2. DeadEnd Colorimetric Apoptosis Detection System was purchased from Promega. Antibodies against procaspase-3 and -9, Bax, Bcl-xL, and poly(ADP-ribose) polymerase (PARP) were purchased from Cell Signaling Technology. Antibodies to E-cadherin, N-cadherin, and β-actin were purchased from Santa Cruz Biotechnology. The Cleaved Caspase-3 [Asp 175] Immunohistochemistry kit was purchased from Cell Signaling Technology. All other chemicals were either reagent or tissue culture grade.
In vitro cytotoxicity studies. The cancer cell lines (A549 and H460) were plated in 96-well microtiter plates at a density of 1 × 106 per well and allowed to incubate overnight. The cells were treated with various dilutions of docetaxel in the presence or absence of C-DIMs (3, 5, and 7 μmol/L). The plates were incubated for 72 h at 37 ± 0.2°C in a 5% CO2-jacketed incubator. Cell viability in each treatment group was determined by crystal violet dye assay.
Data analysis for the combination treatments. The interactions between docetaxel and C-DIMs were evaluated by the isobolographic analysis, a dose-oriented geometric method of assessing drug interactions (34). For 50% toxicity, the combination index (CI) values were calculated based on the equation stated below:
CI=(D)1/(Dx)1+(D)2/(Dx)2+α(D)1(D)2/(Dx)1(Dx)2…CI=(D)1/(Dx)1+(D)2/(Dx)2+α(D)1(D)2/(Dx)1(Dx)2…
where (Dx)1 = dose of drug 1 to produce 50% cell kill alone; (D)1 = dose of drug 1 to produce 50% cell kill in combination with (D)2; (Dx)2 = dose of drug 2 to produce 50% cell kill alone; (D)2 = dose of drug 2 to produce 50% cell kill in combination with (D)1; and α = 0 for mutually exclusive or 1 for mutually nonexclusive modes of drug action. CI > 1.3 indicates antagonism, CI = 1.1 to 1.3 moderate antagonism, CI = 0.9 to 1.1 additive effect, CI = 0.8 to 0.9 slight synergism, CI = 0.6 to 0.8 moderate synergism, CI = 0.4 to 0.6 synergism, and CI = 0.2 to 0.4 strong synergism.
Terminal deoxynucleotidyl transferase-mediated nick end labeling assay of A549 cells. To detect apoptotic cells, the DeadEnd Colorimetric Apoptosis Detection System (Promega) was used. Cells were plated at a density of 1 × 106 per well in 6-well plates and incubated overnight. Cells were treated with docetaxel (0.01 μmol/L), DIM-C-pPhC6H5 (7.5 μmol/L), or combination. Untreated cells were used as control. After 72 h, cells were fixed in 4% paraformaldehyde and mounted onto slides using Cytospin (Shandon). Briefly, the equilibration buffer was added to slides and incubated for 10 min followed by 5 min incubation in 0.2% Triton-X solution. The slides were washed in PBS and incubated with terminal deoxynucleotidyl transferase enzyme at 37°C for 1 h in a humidified chamber for incorporation of biotinylated nucleotides at the 3′-OH ends of DNA. The slides were incubated in horseradish peroxidase-labeled streptavidin to bind the biotinylated nucleotides followed by detection with stable chromogen DAB. The images on the slides were visualized with an Olympus BX40 light microscope equipped with a computer-controlled digital camera (QImaging) and Imaging software (Q Capture). To quantify the apoptotic cells from terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) assay, 100 cells from 6 random microscopic fields were counted.
Measurement of cleaved PARP levels. A549 cells (1 million) were plated in 10 mL growth medium in 25 cm3 flasks and incubated overnight. Cells were treated with docetaxel (0.01 μmol/L) alone, DIM-C-pPhC6H5 (7.5 μmol/L) alone, and combination. The control cells were untreated. The cells were lysed in radioimmunoprecipitation assay buffer [50 mmol/L Tris-HCl (pH 8.0) with 150 mmol/L NaCl, 1.0% Igepal CA-630 (NP-40), 0.5% sodium deoxycholate, and 0.1% SDS] with protease inhibitor (phenylmethylsulfonyl fluoride). Protein content was measured using BCA Protein Assay Reagent Kit (Pierce). The level of cleaved PARP was estimated using the PARP Cleaved [214/215] ELISA kit (Invitrogen) according to the protocol described in manufacturer’s instructions. Briefly, each lysate (20 μg) was incubated with anti-cleaved PARP (detection antibody) for 3 h. After washing, each sample in a well was incubated with a secondary antibody, IgG-horseradish peroxidase solution for 30 min. Stabilized chromagen was added to each well for another 30 min followed by addition of stop solution. The absorbance of each well was read at 450 nm using a microplate reader.
Western blot analysis. Protein was extracted from control and treated cells in radioimmunoprecipitation assay buffer [50 mmol/L Tris-HCl (pH 8.0) with 150 mmol/L NaCl, 1.0% Igepal CA-630 (NP-40), 0.5% sodium deoxycholate, and 0.1% SDS] with protease inhibitor (500 mmol/L phenylmethylsulfonyl fluoride). Protein content was measured using BCA Protein Assay Reagent Kit (Pierce). Equal amounts of supernatant protein (50 μg) from the control and different treatments were denatured by boiling for 5 min in SDS sample buffer, separated by 10% SDS-PAGE, and transferred to nitrocellulose membranes for immunoblotting. Membranes were blocked with 5% skim milk in TBS with Tween 20 [10 mmol/L Tris-HCl (pH 7.6), 150 mmol/L NaCl, and 0.5% Tween 20] and probed with antibodies against caspase-3 and -9 (1:1,000), PARP (1:1,000), Bax (1:1,000), Bcl-xL (1:1,000), E-cadherin (1:200), N-cadherin (1:1,000), and β-actin (1:1,000; Santa Cruz Biotechnology). Horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) were used. Enhanced chemiluminescent solution (Pierce) was used for detection.
In vivo orthotopic lung tumor model. Female, 6-week-old, athymic nu/nu mice were purchased from Harlan. The mice were housed and maintained in specific pathogen-free conditions in a facility approved by the American Association for Accreditation of Laboratory Animal Care. Food and water were provided ad libitum to the animals in standard cages. All experiments were done in accordance with the guidelines of the Institutional Animal Care and Use Committee, Florida A&M University.
Mice were anesthetized and a 5 mm skin incision was made to the left chest, ∼5 mm below the scapula. Hamilton syringes (1 mL) with 28-guage hypodermic needles were used to inject the cell inoculum through the sixth intercostal space into the left lung. The needle was quickly advanced to a depth of 3 mm and quickly removed after the injection of the A549 cells (1 × 106 per mouse) suspended in 100 μL PBS (pH 7.4) into the lung parenchyma. Only cell suspensions of >90% viability determined by trypan blue exclusion were used. Wounds from the incisions were closed with surgical skin clips. Animals were observed for 45 to 60 min until fully recovered.
The control group received 160 μL vehicle; the second group (n = 8) received docetaxel 10 mg/kg given intravenously on days 14, 18, and 22; the third group (n = 8) was given DIM-C-pPC6H5 40 mg/kg three times a week by oral gavage; and the fourth group (n = 8) received a combination of docetaxel and DIM-C-pPhC6H5. To check for evidence of toxicity, the animals were weighed twice weekly. On day 28, all animals were sacrificed by exposure to a lethal dose of halothane in a desiccator. After dissection and removal of the lungs, the lungs and tumors were washed in sterile PBS and weighed. Weights of orthotopic lung tumors were used for assessment of therapeutic activity of the treatments. For immunohistochemistry, TUNEL, and H&E staining procedures, some of the tumors were fixed in formalin, whereas others were rapidly frozen in liquid nitrogen and stored in −80°C.
Immunohistochemistry and TUNEL of orthotopic lung tumor tissues. Sections prepared from formalin-fixed, paraffin-embedded lung tissues were used for immunohistochemical studies according to the protocol specified in the SignalStain Cleaved Caspase-3 [Asp175] Immunohistochemistry kit (Cell Signaling Technology). Sections were cleared in xylene and hydrated in different concentrations of alcohol. The slides were heated in sodium acetate solution at 95°C for 10 min for antigen retrieval. The slides were washed three times in PBS and incubated with the primary antibody against cleaved caspase-3 overnight at 4°C. Horseradish peroxidase-conjugated secondary antibody was applied to locate the primary antibody. Specimens were stained with Nova Red stain and counterstained with hematoxylin. The presence of brown staining was considered a positive identification for activated caspase-3. The Olympus BX40 light microscope equipped with computer-controlled digital camera and imaging software (Q capture) was used to visualize images on the slides.
For TUNEL assay, formalin-fixed tumor tissues harvested 28 days after tumor implantation were embedded in paraffin and sectioned. DeadEnd Colorimetric Apoptosis Detection System (Promega) was used to detect apoptosis in the tumor sections placed on slides according to the manufacturer’s protocol. Briefly, the equilibration buffer was added to slides and incubated for 10 min followed by 10 min incubation in 20 μg/mL proteinase K solution. For the rest of the assay, the steps mentioned in TUNEL Assay of A549 Cells were repeated.
Statistics. One-way ANOVA followed by Tukey’s multiple comparison test was done to determine the significance of differences among groups. Differences were considered significant in all experiments (*, P < 0.05, significantly different from untreated controls; **, P < 0.05, significantly different from DIM-C-pPC6H5 and docetaxel single treatments; ▪, P < 0.05, significantly different from docetaxel single treatment unless otherwise stated). The statistical analysis was done using GraphPad PRISM version 3.0 software.
Results
Inhibition of cell proliferation by C-DIMs in A549 and H460 cells. Results in Fig. 1 show that DIM-C-pPhC6H5, DIM-C-pPhtBu, and DIM-C-pPhCF3 inhibit proliferation of A549 and H460 cells in a dose-dependent manner. In A549 cells, the IC50 values varied between 8 and 12 μmol/L with the relative potency in the order of DIM-C-pPhtBu > DIM-C-pPhC6H5 > DIM-C-pPhCF3. In H460 cells, the IC50 values varied between 7 and 8 μmol/L with the same relative potency, DIM-C-pPhtBu > DIM-C-pPhC6H5 > DIM-C-pPhCF3.
Fig. 1.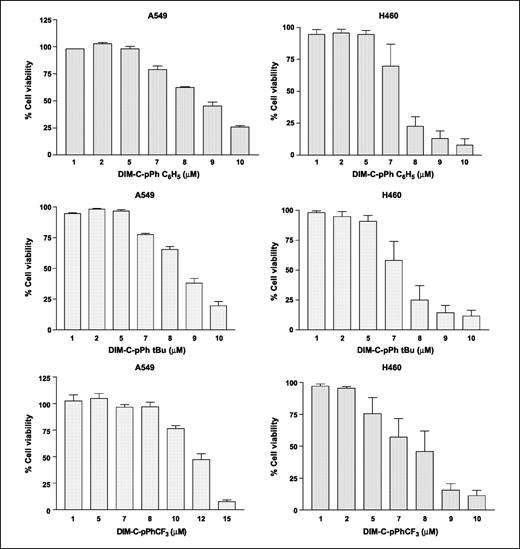 VIEW LARGEDOWNLOAD SLIDE
VIEW LARGEDOWNLOAD SLIDE
In vitro cytotoxicity profiles of C-DIMs in A549 and H460 cells. Cells were treated with C-DIMs and cytotoxicity was determined using the crystal violet dye assay as described in Materials and Methods. Mean ± SE of at least three separate determinations for each treatment group. Dose-dependent inhibition of cell proliferation was noted.
Analysis of the in vitro cytotoxicity of docetaxel and C-DIMs combination. The combined effects of docetaxel and each of the C-DIMs on cell proliferation were evaluated by the isobolographic analysis method. The CI values against both cell lines at three concentrations of DIM-C-pPhC6H5 in combination with docetaxel indicated additive (CI = 0.83-0.99) and synergistic (CI = 0.36-0.75) interactions.
Induction of apoptotic DNA fragmentation in A549 cells.In situ deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) was used to identify 3′-hydroxyl end of fragmented DNA, which is one of the biochemical markers of apoptosis. Figure 2A shows that apoptosis is induced in A549 cells following treatment with 0.01 μmol/L docetaxel, 7.5 μmol/L DIM-C-pPhC6H5, or their combination. Positive TUNEL staining was observed in some A549 cells at 48 h and the highest percentage (Fig. 2B) of apoptotic cells was observed in the cells treated with the combination of docetaxel + C-DIM. Positive TUNEL staining was minimal in control (untreated) cells. DIM-C-pPhC6H5 + docetaxel treatment led to apoptosis in 49% of cells compared with 18% and 30% in cells treated with DIM-C-pPhC6H5 and docetaxel alone, respectively, for 48 h. All treatments were significantly different from control (*, P < 0.001). Docetaxel treatment was significantly different from treatment with the combination of docetaxel and DIM-C-pPhC6H5 (▪, P < 0.001).
Fig. 2.<img src=”https://aacr.silverchair-cdn.com/aacr/content_public/journal/clincancerres/15/2/10.1158_1078-0432.ccr-08-1558/3/m_543fig02g.jpeg?Expires=1659679151&Signature=ZPCXSdwYwInZZimV5e48FTKeEhXG0WSpp-5VUOPbbPl4-XEXd3juvZ2rH18b0~7SWk2vVXPg5CLNuMTE8Px9RIXTT1RptTbk4OY5QFxv~Oc7cGEKISx99fcsly2bN10gfk1OmbZOjY8VO~BIZ7FnXIGZpsMDpj6PIJsHYphmdwCab0I-3ZbdbPuLYJrePeoBKoKwBwMZva5kA~akSsumBqm7a1RRAsYPAw6KI8AXsM5O4aDCGVAvVD2-hcxxHiTaCYhwpsRZtboUA8fhfMP1N~sudr6tgLcPUhkr~JB26R9JlUAUuh7dQ-cfg1dY-oPhRjlj40TOFCkhLHjcx3N5hA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA” alt=”Fig. 2. Induction of DNA fragmentation in A549 cells. A, representative micrographs of A549 cells stained with TUNEL after 48 h with 0.01 μmol/L docetaxel, 7.5 μmol/L DIM-C-pPhC6H5, 0.01 μmol/L and docetaxel + 7.5 μmol/L DIM-C-pPhC6H5. DNA fragmentation is indicated by positive staining (arrows). Control cells were untreated. Micron bar, 10 μm. Arrows, apoptotic cells. Original magnification ×100. B, quantitation of apoptotic cells from TUNEL assay. Cells were quantitated by counting 100 cells from six random microscopic fields. Highest percentage of apoptotic cells was observed in the cells treated with the docetaxel + DIM-C-pPhC6H5. Columns, mean (n = 6); bars, SD. One-way ANOVA followed by post Tukey test was used for statistical analysis to compare control and treated groups. All treatments were significantly different from control (*, P < 0.001). Docetaxel treatment was significantly different from treatment with the combination (▪, P VIEW LARGEDOWNLOAD SLIDE
Induction of DNA fragmentation in A549 cells. A, representative micrographs of A549 cells stained with TUNEL after 48 h with 0.01 μmol/L docetaxel, 7.5 μmol/L DIM-C-pPhC6H5, 0.01 μmol/L and docetaxel + 7.5 μmol/L DIM-C-pPhC6H5. DNA fragmentation is indicated by positive staining (arrows). Control cells were untreated. Micron bar, 10 μm. Arrows, apoptotic cells. Original magnification ×100. B, quantitation of apoptotic cells from TUNEL assay. Cells were quantitated by counting 100 cells from six random microscopic fields. Highest percentage of apoptotic cells was observed in the cells treated with the docetaxel + DIM-C-pPhC6H5. Columns, mean (n = 6); bars, SD. One-way ANOVA followed by post Tukey test was used for statistical analysis to compare control and treated groups. All treatments were significantly different from control (*, P < 0.001). Docetaxel treatment was significantly different from treatment with the combination (▪, P < 0.001).
Increased levels of cleaved PARP in A549 cells. At 48 h, docetaxel alone (*, P < 0.001) and the combination (P < 0.001) treatments significantly increased concentration of cleaved PARP (6-fold) compared with control. Treatment with DIM-C-pPhC6H5 alone (*, P < 0.001) and docetaxel + DIM-C-pPhC6H5 (*, P < 0.001) for 72 h increased PARP cleavage to 1.5- and 2-fold, respectively (data not shown), compared with control. The combination significantly activated PARP cleavage compared with docetaxel or DIM-C-pPhC6H5 alone after treatment for 72 h. It is evident that the apoptotic activity of docetaxel was marginal after 72 h relative to that observed at 48 h.
Effects of treatments on proapoptotic proteins and epithelial-to-mesenchymal transition markers in A549 cells. To further study the role of apoptosis induced by docetaxel and DIM-C-pPhC6H5, we evaluated expression of PARP, procaspase-3, procaspase-9, Bax, and antiapoptotic Bcl-xL proteins by Western blotting done on whole-cell lysates from control and treated cells. Results (Fig. 3A-C) show that the combination significantly (P < 0.001) increased cleaved levels of PARP protein by 5-fold compared with control. After 72 h, the combination, docetaxel, and DIM-C-pPhC6H5 decreased procaspase-3 expression to 0.30 (P < 0.001), 0.50 (P < 0.001), and 0.80-fold (P < 0.001), respectively, compared with controls. The drug combination decreased procaspase-9 expression to 0.6 (P < 0.01) after treatment for 72 h and docetaxel + DIM-C-pPhC6H5 decreased Bcl-xL expression to 0.02-fold (P < 0.001), 0.80-fold (P < 0.001), and 0.40-fold (P < 0.001), respectively, compared with controls. At 72 h, the combination increased Bax expression by 3-fold (P < 0.001) compared with a 1.5-fold (P < 0.05) increase by docetaxel or DIM-C-pPhC6H5 alone. Two epithelial-to-mesenchymal transition (EMT) markers E-cadherin and N-cadherin were also studied by Western blotting. As shown in Fig. 4, after 72 h, the combination decreased E-cadherin expression to 0.50-fold (P < 0.05) compared with control. In contrast, the drug combination, docetaxel, and DIM-C-pPhC6H5 alone increased N-cadherin expression 18-fold (P < 0.001), 6.5-fold (P < 0.01), and 3.0-fold, respectively, compared with controls. β-Actin protein was used as a loading control.
Fig. 3.<img src=”https://aacr.silverchair-cdn.com/aacr/content_public/journal/clincancerres/15/2/10.1158_1078-0432.ccr-08-1558/3/m_543fig03g.jpeg?Expires=1659679151&Signature=W6qutlI-xFuXiH6b8X1WA8s1aVLtdXJxTT2EjOZcqqBdbpftfM6zUYFi6c6BUCOuscOix5LBXYLlmiKA3r2g9rZ8sytQhezFOfO-yKepTvhOYt~tYg1M63hBan2nSKKc0yQ9obETq8RQ9OFJtZk172DT0~nEkXHIfR0Qi86O8j8wxYJUMH2f7SoHNEHVmhDdLdvW5xWPwZU4VnUefrm7Ru~8X7cudYi8s78Ph2yx2949leVUow9~JZdFRXMfpjSkgpTeNPB57307XpvpgAt9aphJmCFw0Tu47GQEnKa-lJhUeUCMNOuDEEZKaqhn3GPq4ecpa-DOjVBkGwCYvGnRpg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA” alt=”Fig. 3. Expression of apoptosis proteins. A549 cells were treated with docetaxel (0.01 μmol/L), DIM-C-pPhC6H5 (7.5 μmol/L), and docetaxel (0.01 μmol/L) + DIM-C-pPhC6H5 (7.5 μmol/L) for 48 and 72 h and whole-cell lysates were analyzed by Western blotting for PARP, procaspase-3 and -9, and Bcl-xL (A) and Bax (B) protein expression. Lane 1, untreated control cells; lane 2, docetaxel (0.01 μmol/L); lane 3, DIM-C-pPhC6H5 (7.5 μmol/L); lane 4, docetaxel (0.01 μmol/L) + DIM-C-pPhC6H5 (7.5 μmol/L). β-Actin protein acts as a loading control. Similar results were observed in replicate experiments. C, quantitation of apoptosis protein expression. Protein expression levels (relative to β-actin) were determined. Mean ± SE of three replicate determinations. *, P < 0.05, significantly different from untreated controls; **, P < 0.05, significantly different from DIM-C-pPC6H5 and docetaxel single treatments; ▪, P VIEW LARGEDOWNLOAD SLIDE
Expression of apoptosis proteins. A549 cells were treated with docetaxel (0.01 μmol/L), DIM-C-pPhC6H5 (7.5 μmol/L), and docetaxel (0.01 μmol/L) + DIM-C-pPhC6H5 (7.5 μmol/L) for 48 and 72 h and whole-cell lysates were analyzed by Western blotting for PARP, procaspase-3 and -9, and Bcl-xL (A) and Bax (B) protein expression. Lane 1, untreated control cells; lane 2, docetaxel (0.01 μmol/L); lane 3, DIM-C-pPhC6H5 (7.5 μmol/L); lane 4, docetaxel (0.01 μmol/L) + DIM-C-pPhC6H5 (7.5 μmol/L). β-Actin protein acts as a loading control. Similar results were observed in replicate experiments. C, quantitation of apoptosis protein expression. Protein expression levels (relative to β-actin) were determined. Mean ± SE of three replicate determinations. *, P < 0.05, significantly different from untreated controls; **, P < 0.05, significantly different from DIM-C-pPC6H5 and docetaxel single treatments; ▪, P < 0.05, significantly different from docetaxel single treatment.
Fig. 4.<img src=”https://aacr.silverchair-cdn.com/aacr/content_public/journal/clincancerres/15/2/10.1158_1078-0432.ccr-08-1558/3/m_543fig04g.jpeg?Expires=1659679151&Signature=wOb4sCTIr9TDbtI0s7TvS2ngXtqAarpek9SXYMGjPpFzOiItMQbIYh3hp96I8ScRMd4Jul40jErKy~lqHKQwMIfaXyCtxVbEFqaSBW5lUno4jUtHWR69~sXKEcwq-4879KOWuyBbugP9pI~Jy8ovL9g7FtUFlYjNeVu5ICrJVDlgUA0PDCp6bgKtsgvluqGUTzjdPRWpwuq-NbAnNlRvUE2GW69IrCjItrQIVEfYUoG~4TKD~r0ne9Xz~BDu3RiVmqFKeiXlYgnkACFr3~pFcRywXtmdcTs3dshtxliMDO-TkMV7JAGzRcAVvRDc8ESxBDn~Vo4sLVciBSlTeazzuA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA” alt=”Fig. 4. Expression of EMT proteins. A, A549 cells were treated with docetaxel (0.01 μmol/L), DIM-C-pPhC6H5 (7.5 μmol/L), and docetaxel (0.01 μmol/L) + DIM-C-pPhC6H5 (7.5 μmol/L) for 72 h and whole-cell lysates were analyzed by Western blotting for E-cadherin and N-cadherin protein expression. Lane 1, untreated control cells; lane 2, docetaxel (0.01 μmol/L); lane 3, DIM-C-pPhC6H5 (7.5 μmol/L); lane 4, docetaxel (0.01 μmol/L) + DIM-C-pPhC6H5 (7.5 μmol/L). β-Actin protein acts as a loading control. Similar results were observed in replicate experiments. B, quantitation of EMT protein expression. Protein expression levels (relative to β-actin) were determined. Mean ± SE of three replicate determinations. *, P < 0.05, significantly different from untreated controls; **, P < 0.05, significantly different from DIM-C-pPC6H5 and docetaxel single treatments; ▪, P VIEW LARGEDOWNLOAD SLIDE
Expression of EMT proteins. A, A549 cells were treated with docetaxel (0.01 μmol/L), DIM-C-pPhC6H5 (7.5 μmol/L), and docetaxel (0.01 μmol/L) + DIM-C-pPhC6H5 (7.5 μmol/L) for 72 h and whole-cell lysates were analyzed by Western blotting for E-cadherin and N-cadherin protein expression. Lane 1, untreated control cells; lane 2, docetaxel (0.01 μmol/L); lane 3, DIM-C-pPhC6H5 (7.5 μmol/L); lane 4, docetaxel (0.01 μmol/L) + DIM-C-pPhC6H5 (7.5 μmol/L). β-Actin protein acts as a loading control. Similar results were observed in replicate experiments. B, quantitation of EMT protein expression. Protein expression levels (relative to β-actin) were determined. Mean ± SE of three replicate determinations. *, P < 0.05, significantly different from untreated controls; **, P < 0.05, significantly different from DIM-C-pPC6H5 and docetaxel single treatments; ▪, P < 0.05, significantly different from docetaxel single treatment.
Antitumor effects of docetaxel + DIM-C-pPhC6H5 in A549 orthotopic lung tumors. The in vivo anticancer activity of DIM-C-pPhC6H5 alone and in combination with docetaxel was further investigated in female athymic nude mice bearing A549 orthotopic lung tumors. Results of pilot studies established that implantation of 1 × 106 A549 cells in the lungs of nude mice generated fairly uniform tumors within 7 to 14 days. Treatment was started 7 days after tumor implantation and continued for a total of 21 days. The results (Fig. 5) show that lung tumor weights were significantly decreased after treatment with docetaxel (10 mg/kg given intravenously; P < 0.001), DIM-C-pPhC6H5 (40 mg/kg given orally; P < 0.01), or docetaxel + DIM-C-pPhC6H5 (P < 0.001) compared with controls. It is evident that docetaxel + DIM-C-pPhC6H5 was most effective inhibitor of lung tumor growth compared with docetaxel or DIM-C-pPhC6H5 treatments. At the end of the study period (28 days), there was a 57% reduction in lung weight after treatment with docetaxel + DIM-C-pPhC6H5 compared with a 39% or 22% decrease after treatment with docetaxel or DIM-C-pPhC6H5 alone, respectively. We did not observe any weight loss or other signs of toxicity in the mice treated with DIM-C-pPhC6H5 (data not shown).
Fig. 5.<img src=”https://aacr.silverchair-cdn.com/aacr/content_public/journal/clincancerres/15/2/10.1158_1078-0432.ccr-08-1558/3/m_543fig05l.jpeg?Expires=1659679151&Signature=pD3-yB4FrDlE0nDw3uYc2HHPizw6cOrHCySHm1XYqjpqOA03A0L~56Q5kMij7g2Rm3jxGe1uCCRLs3wwHhVoI0jhOEx027lPwlFiaSud3DNWkYANncOaBJyUbMDUrO-kaLIVhW6ILLJ2ouI0O8tFn4HP7afhz2a5KjoAbdtq~q1s3LZ4o0OAm85KVLdWk7sQOy6CzZADLlsE4L~kDLnZnVlztXj0~-yFBbt9ALPeFtU~4vICW10sWyjkPZuO4tNa7036A2U9In17CxJMnLSRRrueu5K~OWY6Q05mFog-YR2wRPiFaBWX~xLNkDcGW5gMe-0xoLXVgaLWHh3M6Nt~IA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA” alt=”Fig. 5. Effect of DIM-C-pPhC6H5 and docetaxel treatments on mice lung weights. Female nude mice with orthotopic lung tumors received various treatments for 28 d starting on day 7 following tumor implantation. The mice were treated with docetaxel 10 mg/kg given intravenously (days 14, 18, and 22), DIM-C-pPhC6H5 40 mg/kg given orally (3 times a week), and docetaxel + DIM-C-pPhC6H5. Control group received vehicle only. Mean ± SE (n = 8). This experiment was repeated twice. *, P < 0.05, significantly different from untreated controls; **, P VIEW LARGEDOWNLOAD SLIDE
Effect of DIM-C-pPhC6H5 and docetaxel treatments on mice lung weights. Female nude mice with orthotopic lung tumors received various treatments for 28 d starting on day 7 following tumor implantation. The mice were treated with docetaxel 10 mg/kg given intravenously (days 14, 18, and 22), DIM-C-pPhC6H5 40 mg/kg given orally (3 times a week), and docetaxel + DIM-C-pPhC6H5. Control group received vehicle only. Mean ± SE (n = 8). This experiment was repeated twice. *, P < 0.05, significantly different from untreated controls; **, P < 0.05, significantly different from DIM-C-pPC6H5 and docetaxel single treatments.
Cleaved caspase-3 expression in A549 orthotopic lung tumors. Expression of cleaved caspase-3 was also investigated in the control and treated tumors by immunohistochemistry analysis. Docetaxel (10 mg/kg given intravenously), DIM-C-pPhC6H5 (40 mg/kg given orally), and docetaxel + DIM-C-pPhC6H5 induced caspase-3 expression in tumors compared with control tumors (Fig. 6A).
Fig. 6.<img src=”https://aacr.silverchair-cdn.com/aacr/content_public/journal/clincancerres/15/2/10.1158_1078-0432.ccr-08-1558/3/m_543fig06g.jpeg?Expires=1659679151&Signature=0XgEXhmFwr3pJkYo5t1xR~2-jC0w87xJEiBvJJQS1kbqUbfj4EjzOGxHM99Tv-krJnQ5r~IMUjXHFBMq2evlQhlYg7mnyHkeutkywJObvRGv-032m4H8SshUvprDRDALdYzbScnFU-Wht2Eram8e9besI~S2Jy-~W-ndXwyrfg-gzwMJA0bsqW-1LPkzCaEV59e3bNxRRki0fH8WXnq536J~7GTmalo8Dymz1PppJExtKPfWhZFvDPr9HCH9N5DJbUCCOdr7GKXTzLUNubtcokeAIvhDFx9oMzKIS1uLUhNdIIoJ6Vq563m-Xi3XGZo7kUxgJDVbqFxLkCvX6IPYzw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA” alt=”Fig. 6. Expression of cleaved caspase-3 and induction of DNA fragmentation in A549 tumors. A, immunohistochemical staining of orthotopic A549 lung tumor tissues for cleaved caspase-3 expression. Lungs were dissected from mice on day 28, fixed in 10% formalin, paraffin embedded, and sectioned. Sections were stained using the cleaved caspase-3 (Asp 175) immunohistochemistry kit as described in Materials and Methods. Cells showing caspase-3 cleavage are stained. Original magnification ×100. B, TUNEL staining of orthotopic A549 lung tumor tissues. Lungs were dissected from mice on day 28, fixed in 10% formalin, paraffin embedded, and sectioned. Sections were stained using the DeadEnd colorimetric kit as described in Materials and Methods. The apoptotic tumor cells are stained. Original magnification ×100. C, quantitation of apoptotic cells from TUNEL staining. Percentages of TUNEL-positive cells were quantitated by counting 100 cells from six random microscopic fields each in three tumor samples from different treatment groups. Columns, mean (n = 6); bars, SE. P VIEW LARGEDOWNLOAD SLIDE
Expression of cleaved caspase-3 and induction of DNA fragmentation in A549 tumors. A, immunohistochemical staining of orthotopic A549 lung tumor tissues for cleaved caspase-3 expression. Lungs were dissected from mice on day 28, fixed in 10% formalin, paraffin embedded, and sectioned. Sections were stained using the cleaved caspase-3 (Asp 175) immunohistochemistry kit as described in Materials and Methods. Cells showing caspase-3 cleavage are stained. Original magnification ×100. B, TUNEL staining of orthotopic A549 lung tumor tissues. Lungs were dissected from mice on day 28, fixed in 10% formalin, paraffin embedded, and sectioned. Sections were stained using the DeadEnd colorimetric kit as described in Materials and Methods. The apoptotic tumor cells are stained. Original magnification ×100. C, quantitation of apoptotic cells from TUNEL staining. Percentages of TUNEL-positive cells were quantitated by counting 100 cells from six random microscopic fields each in three tumor samples from different treatment groups. Columns, mean (n = 6); bars, SE. P < 0.05: *, significantly different from untreated controls; **, significantly different from DIM-C-pPC6H5 and docetaxel single treatments.
Effects of docetaxel and DIM-C-pPhC6H5 on DNA fragmentation in A549 orthotopic lung tumors. To further investigate the role of apoptosis, tumor sections were harvested at the end of the study and stained with TUNEL for detection of DNA fragmentation (Fig. 6B). Single-agent therapy with either DIM-C-pPhC6H5 or docetaxel induced DNA fragmentation (brown staining) that was further increased by combination therapy. DIM-C-pPhC6H5 + docetaxel treatment led to apoptosis in 43% of the tumor cells, whereas DIM-C-pPhC6H5 and docetaxel alone induced apoptosis in 22% and 29% of the tumor cells, respectively (Fig. 5C). All treatments were significantly different from control (*, P < 0.001). Docetaxel treatment was significantly different from treatment with the combination of docetaxel and DIM-C-pPhC6H5 (▪, P < 0.001).
Discussion
The overexpression of PPARγ in cancer cells and tumors (8, 11) has generated considerable interest in developing new potent and pharmacologically safe anticancer drugs that target PPARγ and activate growth inhibitory and proapoptotic pathways (35). Several reports have established that PPARγ agonists are potent anticancer drugs, but surprisingly many of their effects are mediated by receptor-independent mechanisms (21, 25, 36). Based on these reports, we evaluated the potential of DIM-C-pPhC6H5 in potentiating antitumor activity of docetaxel in vitro and in vivo in A549 orthotopic lung tumors and to elucidate pathways that contribute to the anticarcinogenic responses.
In this study, we showed that C-DIMs enhanced the in vitro cytotoxicity of docetaxel in A549 and H460 cells. The IC50 values for each compound in both lung cancer cell lines ranged between 7 and 12 μmol/L. Our results are similar to those observed for the C-DIMs in other cancer cell lines where there was concentration-dependent inhibition of cell growth at concentrations ≥5 μmol/L (22–24). C-DIMs inhibited growth of HT-29, HCT-15, SW480, and RKO colon cancer cell lines at IC50 values between 1 and 10 μmol/L (22). C-DIMs inhibited the growth of KU7 and 253J-BV bladder cancer cells at the IC50 values of 5 to 10 and 1 to 5 μmol/L, respectively (23). A recent study reported that the growth-inhibitory and apoptotic effects of C-DIMs against LNCaP cells were correlated with decreased cyclin D1 expression, induction of p27 (concentration-dependent) and nonsteroidal anti-inflammatory drug activated gene-1, and down-regulation of caveolin-1 (24).
In the present investigation, isobolographic analysis of the data showed that the C-DIMs enhance the cytotoxicity of docetaxel in A549 and H460 human NSCLC cells and these effects were both additive and synergistic. We recently reported that the CI values in both cell lines using different concentrations of 15-deoxy-Δ12,14-prostaglandin J2 in combination with docetaxel were <1.0 indicative of synergistic interactions (33). Our results are consistent with the work of Menendez et al. who showed by isobolographic analysis that the cytotoxicity of anticancer drugs such as doxorubicin in breast cancer cell lines was synergistically or additively modified by fatty acids (34). Another study showed synergistic interactions for sulindac sulfide, exisulind, and nordihydroguaiaretic acid with paclitaxel, cisplatin, and 13-cis-retinoic acid in A549, H460, and SHP77 human lung cancer cell lines (37). Exisulind at the highest concentration was synergistic with paclitaxel (CI = 0.4-0.8), and additive (CI = 0.9-1.1) or synergistic (CI = 0.6-0.9) effects were observed with cisplatin in the three cell lines tested. Combinations of 13-cis-retinoic acid with paclitaxel showed strong synergy (CI = 0.2-0.4) in NSCLC cells.
DIM in combination with paclitaxel inhibited cell growth by 74 ± 1.07% compared with 42 ± 3.26% and 62 ± 1.12% by DIM and paclitaxel alone, respectively (3). We selected DIM-C-pPhC6H5 for further investigation in NSCLC cells because of our interest in following the work of Qin et al., who reported that the DIM-C-pPhC6H5 inhibited carcinogen-induced rat mammary tumor growth at a low dose of 1 mg/kg administered every other day (13). We hypothesized that DIM-C-pPhC6H5 induces apoptosis in lung cancer cells by different pathways and that the combined treatment would be synergistic.
To study the mechanism involved in the enhanced cytotoxicity of docetaxel by the selected C-DIM (DIM-C-pPhC6H5), we evaluated the potential of each agent alone and in combination in inducing apoptosis in A549 cells after treatment for 48 h. The TUNEL assay results showed induction of apoptosis (Fig. 2) and these results were similar to those observed with 15-deoxy-Δ12,14-prostaglandin J2 and docetaxel where significant (P < 0.001) positive TUNEL staining in A549 cells was observed after 48 h treatment with the combination of both drugs. Compared with docetaxel alone, 15-deoxy-Δ12,14-prostaglandin J2 + docetaxel induced 1.5-fold increase in apoptosis (33). McGuire et al. also reported that DIM + paclitaxel treatment induced apoptosis in 34.8% of treated cells, whereas DIM and paclitaxel induced apoptosis in 6.59% and 7.39%, respectively, in HER-2/neu-overexpressing human breast cancer cells (3).
Because data from the TUNEL assay provided evidence for enhanced induction of apoptosis with docetaxel + DIM-C-pPhC6H5 combination, we examined their effects on several proapoptotic responses. Figure 3 indicates that simultaneous cotreatment of docetaxel and DIM-C-pPhC6H5 decreased expression of procaspase-3, procaspase-9, and Bcl-xL after 72 h and increased expression of Bax after 48 h compared with single-agent treatment and controls. Furthermore, cleaved PARP, an early marker of apoptosis was highest in the combination treatment group after 72 h compared with treatment with the compounds alone. These results suggest that apoptosis may be mediated through the mitochondrial pathway via down-regulation of antiapoptotic Bcl-xL and up-regulation of proapoptotic Bax. Therefore, we reasoned that cell damage resulting from treatment altered Bax/Bcl-xL ratios resulting in activation of caspase-9 and subsequent activation of downstream proapoptotic pathways.
Studies in other cell lines show that compounds including DIM, parthenolide, a sesquiterpene lactone, and bortezomib enhance cytotoxicity and cell death in combination with taxanes (3, 32, 38). McGuire et al. also showed that DIM in combination with paclitaxel led to synergistic apoptotic response mediated by the mitochondrial pathway (Bcl-2/PARP) in HER-2/neu human breast cancer cells. In these cells, expression of Bcl-2 was significantly reduced in the combination treatments, but Bax protein expression remained unchanged, whereas DIM showed no effect on Bax. PARP cleavage was significantly increased in cells cotreated with DIM and paclitaxel compared with treatment with the compounds alone (3). We also examined how markers of EMT are affected by C-DIM and docetaxel treatment and observed the loss of an epithelial cell marker (E-cadherin) and induction of a marker for mesenchyme (N-cadherin), suggesting occurrence of an EMT (Fig. 4). Molecular mechanisms of EMT induction by C-DIMs and docetaxel need to be further explored in lung cancer as an important contributor of their anticarcinogenic activity.
Having established the effectiveness of the combination treatment in vitro, we next evaluated the effects of docetaxel + DIM-C-pPhC6H5 on growth of established A549 orthotopic lung tumors in vivo. For the first time, we show that DIM-C-pPhC6H5 exhibits antitumorigenic activity alone against orthotopic lung tumors in mice. The combination of DIM-C-pPhC6H5 + docetaxel caused a significant (P < 0.01) decrease in lung tumor weights compared with controls or either agent alone (Fig. 5). We would expect this treatment to be also successful if started later than day 7 post-tumor implantation because previous studies with C-DIMs showed tumor growth inhibition when these compounds were administered after formation of palpable tumors in xenograft studies (20, 22, 24). Our study and others (32, 33) have shown evidence of tumor growth inhibition when docetaxel treatment was initiated 14 days post-tumor implantation after tumors were fairly established. Several studies show that enhanced tumor growth inhibition can be achieved by combining docetaxel with other agents as opposed to treating with either agent alone (29–33). Moreover, the combination of parthenolide and docetaxel was more effective than the compounds alone in the MDA-MB-231 cell-derived xenograft metastasis model of breast cancer (32) and these results are similar to those observed in this study on lung cancer models.
We also investigated the effects of docetaxel, DIM-C-pPhC6H5, and their combination on apoptosis in tumors by determining caspase-3 expression and DNA fragmentation. Increased expression of cleaved caspase-3 (Fig. 6A) and increased number of apoptotic cells (Fig. 6B and C) were observed in tumors treated with the combination compared with tumors treated with docetaxel or DIM-C-pPhC6H5 alone. These in vivo responses correlated with induced DNA fragmentation and activation of caspase-3 as observed in A549 cells treated with the same compounds (Figs. 2A and 3). Our result supports the work of Chintharlapalli et al. in which DIM-C-pPhC6H5 at a dose of 40 mg/kg/d significantly induced caspase-3 expression compared with control tumors (21). Consistent with our data, it has been reported that combination therapy with bortezomib + docetaxel led to significant (P < 0.05) increases in percentage of apoptotic cells in L3.6pl and MiaPaCa-2 pancreatic tumor xenografts compared with therapy with either drug alone (30).
In conclusion, results of this study show that the combination of docetaxel with DIM-C-pPhC6H5 additively or synergistically enhances the in vitro and in vivo anticancer activity of docetaxel. The enhanced effect may be associated mitochondrial-driven apoptosis involving activation of procaspase-3 and -9, decreased expression of Bcl-xL, and increased expression of Bax and cleaved PARP. Combination therapy also induced apoptosis in the orthotopic tumors and this was also associated with increased expression of cleaved caspase-3. To gain more insights on the mechanisms by which these drugs work together to inhibit growth of cancer cells and tumors, other nonapoptotic signaling pathways including angiogenesis need to be investigated and these studies are in progress.
Disclosure of Potential Conflicts of Interest
S. Safe, consultant, Plantacor, a company that has licensed C-DIM compounds to Texas A & M University.
Grant support: NIH RCMI award G12RR03020-11 and grants CA108718 and CA112337 (S. Safe).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
1American Cancer Society. Cancer Facts and Figures 2006. Atlanta: American Cancer Society; 2006.2Whitehead CM, Earle KA, Fetter J, et al. Exisulind-induced apoptosis in a non-small cell lung cancer orthotopic lung tumor model augments docetaxel treatment and contributes to increased survival. Mol Cancer Ther 2003;2:479–88.3McGuire KP, Ngoubilly N, Neavyn M, Lanza-Jacoby S. 3,3′-Diindolylmethane and paclitaxel act synergistically to promote apoptosis in HER2/Neu human breast cancer cells. J Surg Res 2006;132:208–13.4Copland JA, Marlow LA, Kurakata S, et al. Novel high-affinity PPARγ agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene 2006;25:2304–17.5Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83:835–9.6Chinetti G, Griglio S, Antonucci M, et al. Activation of proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. J Biol Chem 1998;273:25573–80.7Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-Δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 1995;83:803–12.8Keshamouni VG, Reddy RC, Arenberg DA, et al. Peroxisome proliferator-activated receptor-γ activation inhibits tumor progression in non-small-cell lung cancer. Oncogene 2004;23:100–8.9Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell 1995;83:813–9.10Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J Biol Chem 1995;270:12953–6.11Chang TH, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor γ in non-small cell lung cancer. Cancer Res 2000;60:1129–38.12Michael LF, Lazar MA, Mendelson CR. Peroxisome proliferator-activated receptor γ1 expression is induced during cyclic adenosine monophosphate-stimulated differentiation of alveolar type II pneumonocytes. Endocrinology 1997;138:3695–703.13Qin C, Morrow D, Stewart J, et al. A new class of peroxisome proliferator-activated receptor γ (PPARγ) agonists that inhibit growth of breast cancer cells: 1,1-bis(3′-indolyl)-1-(p-substituted phenyl)methanes. Mol Cancer Ther 2004;3:247–60.14Elstner E, Muller C, Koshizuka K, et al. Ligands for peroxisome proliferator-activated receptorγ and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A 1998;95:8806–11.15Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. Characteristics of the peroxisome proliferator activated receptor γ (PPARγ) ligand induced apoptosis in colon cancer cells. Gut 2002;50:658–64.16Takahashi N, Okumura T, Motomura W, Fujimoto Y, Kawabata I, Kohgo Y. Activation of PPARγ inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Lett 1999;455:135–9.17Kumagai T, Ikezoe T, Gui D, et al. RWJ-241947 (MCC-555), a unique peroxisome proliferator-activated receptor-γ ligand with antitumor activity against human prostate cancer in vitro and in beige/nude/ X-linked immunodeficient mice and enhancement of apoptosis in myeloma cells induced by arsenic trioxide. Clin Cancer Res 2004;10:1508–20.18Bren-Mattison Y, Van Putten V, Chan D, Winn R, Geraci MW, Nemenoff RA. Peroxisome proliferator-activated receptor-γ (PPARγ) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC). Oncogene 2005;24:1412–22.19Tsubouchi Y, Sano H, Kawahito Y, et al. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-γ agonists through induction of apoptosis. Biochem Biophys Res Commun 2000;270:400–5.20Satoh T, Toyoda M, Hoshino H, et al. Activation of peroxisome proliferator-activated receptor-γ stimulates the growth arrest and DNA-damage inducible 153 gene in non-small cell lung carcinoma cells. Oncogene 2002;21:2171–80.21Chintharlapalli S, Papineni S, Safe S. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit colon cancer cell and tumor growth through PPARγ-dependent and PPARγ-independent pathways. Mol Cancer Ther 2006;5:1362–70.22Chintharlapalli S, Smith R III, Samudio I, Zhang W, Safe S. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes induce peroxisome proliferator-activated receptor γ-mediated growth inhibition, transactivation, and differentiation markers in colon cancer cells. Cancer Res 2004;64:5994–6001.23Kassouf W, Chintharlapalli S, Abdelrahim M, Nelkin G, Safe S, Kamat AM. Inhibition of bladder tumor growth by 1,1-bis(3′-indolyl)-1-(p-substituted phenyl)methanes: a new class of peroxisome proliferator-activated receptor γ agonists. Cancer Res 2006;66:412–8.24Chintharlapalli S, Papineni S, Safe S. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit growth, induce apoptosis, and decrease the androgen receptor in LNCaP prostate cancer cells through peroxisome proliferator-activated receptor γ-independent pathways. Mol Pharmacol 2007;71:558–69.25Vanderlaag K, Su Y, Frankel AE, et al. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit proliferation of estrogen receptor-negative breast cancer cells by activation of multiple pathways. Breast Cancer Res Treat 2008;109:273–83.26Hong J, Samudio I, Liu S, Abdelrahim M, Safe S. Peroxisome proliferator-activated receptor γ-dependent activation of p21 in Panc-28 pancreatic cancer cells involves Sp1 and Sp4 proteins. Endocrinology 2004;145:5774–85.27Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S. 3,3′-Diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis 2006;27:717–28.28Hida T, Kozaki K, Ito H, et al. Significant growth inhibition of human lung cancer cells both in vitro and in vivo by the combined use of a selective cyclooxygenase 2 inhibitor, JTE-522, and conventional anticancer agents. Clin Cancer Res 2002;8:2443–7.29Hida T, Kozaki K, Muramatsu H, et al. Cyclooxygenase-2 inhibitor induces apoptosis and enhances cytotoxicity of various anticancer agents in non-small cell lung cancer cell lines. Clin Cancer Res 2000;6:2006–11.30Nawrocki ST, Sweeney-Gotsch B, Takamori R, McConkey DJ. The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther 2004;3:59–70.31Shaik MS, Chatterjee A, Jackson T, Singh M. Enhancement of antitumor activity of docetaxel by celecoxib in lung tumors. Int J Cancer 2006;118:396–404.32Sweeney CJ, Mehrotra S, Sadaria MR, et al. The sesquiterpene lactone parthenolide in combination with docetaxel reduces metastasis and improves survival in a xenograft model of breast cancer. Mol Cancer Ther 2005;4:1004–12.33Fulzele SV, Chatterjee A, Shaik MS, Jackson T, Ichite N, Singh M. 15-Deoxy-Δ12,14-prostaglandin J2 enhances docetaxel anti-tumor activity against A549 and H460 non-small-cell lung cancer cell lines and xenograft tumors. Anticancer Drugs 2007;18:65–78.34Menendez JA, del Mar Barbacid M, Montero S, et al. Effects of γ-linolenic acid and oleic acid on paclitaxel cytotoxicity in human breast cancer cells. Eur J Cancer 2001;37:402–13.35Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor γ agonists. Lancet Oncol 2004;5:419–29.36Ikezoe T, Miller CW, Kawano S, et al. Mutational analysis of the peroxisome proliferator-activated receptor γ gene in human malignancies. Cancer Res 2001;61:5307–10.37Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA, Jr., Chou TC. Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines. Cancer Res 1999;59:6178–84.38Dong QG, Sclabas GM, Fujioka S, et al. The function of multiple IκB: NF-κB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene 2002;21:6510–9. American Association for Cancer Research